



关于保健食品中黄芪甲苷含量测定的研究
准确测定保健食品中黄芪甲苷的含量对于含黄芪保健食品的质量监控是非常重要的,而保健食品中的植物性成分严重干扰黄芪甲苷的测定。目前研究的多是测定中成药或中药材中的黄芪甲苷的方法,尚没有建立成分复杂的保健食品中黄芪甲苷含量测定的高效分析方法。目前的可见分光光度法、薄层扫描法、荧光分光光度法等难以实现复杂样品的准确、灵敏、稳定检测;色-质联机用因其昂贵价格在推广应用中受到限制。高效液相色谱-紫外检测器为测定黄芪甲苷的常用方法,易普及,但因黄芪甲苷仅具有末端紫外吸收,加上溶剂背景难以消除、保健食品中性质相近物质干扰等影响,结果易受干扰,且响应较低。故本研究拟摸索提高保健食品中黄芪甲苷测定的紫外响应和消除杂质干扰的方法。
1 材料与方法
1.1 仪器和试剂
1.1.1 仪器高效液相色谱仪(Waters 600,二极管阵列检测器),液相色谱(Thermo Surveyer)一质谱(LTQ)联用仪,氮吹仪 (OA-SYS,Organomation Associates,Inc),紫外分光光度计(岛津,Biospee-1601)。
1.1.2 试剂 黄芪甲苷标准品(中国药品生物制品检定所), 苯甲酰氯,氯仿,吡啶,D-101、D-101-I、DM-301大孔吸附树脂净品,Oasis HLB 3cc固相萃取柱。
1.2 方法
保健食品中多含有脂溶性和水溶性杂质,在文献报道和前期实验摸索基础上,样品经甲醇超声提取和乙醚脱脂、水饱和正丁醇萃取、碱洗三步液液萃取初步去除杂质。实验主要对大孔吸附树脂柱层析、固相萃取、衍生化反应条件和HPLC条件进行了筛选。
1.2.1 大孔吸附树脂条件的研究 样品初步处理后多呈粘稠、色深,检测干扰较大,分析样品中还含有一些性质和黄芪甲苷相近的物质未能去除,如黄酮、其他皂甙类物质。大孔吸 附树脂常用于皂甙类物质的除杂,因此试验选择D-101、D-101-I、DM-301大孔吸附树脂进行柱层析。97μg/ml黄芪甲苷标准溶液过柱,选用多个浓度乙醇和水作为淋洗溶剂,每种条件下依次收集10ml,共收集5次,测定流出液体中黄芪甲苷含量,筛选能最大程度洗脱黄芪甲苷溶剂的浓度和体积,以及不能洗脱黄芪甲苷的浓度和体积。通过加标回收试验考察D-101、D-101-I、DM-301对除杂效果和黄芪甲苷回收率的影响,进而选择最佳型号大孔树脂。
1.2.2 固相萃取条件的研究 大孔树脂分离出的一般为总皂苷类物质,为将黄芪甲苷和其他皂甙类物质分离、富集,选择Oasis HLB 3cc型固相萃取小柱进行固相萃取。试验以97μg/ml黄芪甲苷标准溶液对水和多个浓度的甲醇、乙醇、乙腈淋洗和洗脱溶液进行筛选。
1.2.3 衍生化反应条件的研究 试验发现经上述前处理后 色谱峰仍有干扰,分析是黄芪甲苷在近紫外区仅200nm末端有吸收,很多其他物质在此波长范围内均有吸收。故考虑在其分子上连接发光基团,使紫外吸收波长红移,以此消除干扰。
1.2.3.1 衍生化反应条件的筛选 试验选择苯甲酰氯和三氯甲烷在毗啶溶液中将黄芪甲苷衍生为苯甲酰黄芪甲苷,对反应时间、反应温度、吡啶用量、苯甲酰氯用量四个反应条件按L16—4—5正交设计进行测定,对黄芪甲苷标准溶液进行衍生化,将测得的衍生化产物量和未衍生黄芪甲苷量进行比较,计算回收率,筛选最佳反应条件。
1.2.3.2 衍生化产物的检测波长的测定 将浓度为160μg/ml的黄芪甲苷标准品衍生物显色体系,以试剂为空白,进行波长扫描,确定衍生化产物的检测波长。
1.2.3.3 衍生化产物的质谱验证和稳定性测试黄芪甲苷衍生化产物用液相一电喷雾质谱法进行衍生化产物检测,并测定了衍生化产物在4℃下保存1小时、5小时、l周、1个月、2 个月、4个月时的量,以考察稳定性。
1.2.4 高效液相色谱条件的选择 试验对Kromasil C8 (250mm×4.6mm,5μm)、Nova-Pak C18(150mm x 3.9mm,5μm )、 AccQ Tag TM C18 (150mm×3.9mm,5μm)3种色谱柱和90% 甲醇-4%四氢呋喃-6%水、90%甲醇-4%四氢呋喃-6%水-0.4%三乙胺、90%甲醇-10%水、100% 乙腈4种流动相进行了筛选;流速:1ml/min;柱温:25℃ ;检测波长:230nm。
2 结果
2.1 大孔吸附树脂柱层析条件的确定
结果显示,D-101、D-101-I、DM-301不能洗脱下黄芪甲苷的乙醇最高浓度分别为20% 、30%、30% ,完全洗脱下黄芪甲苷的乙醇最低浓度(所需体积)分别为90% (30ml)、70% (20ml)、80%(20ml)。
三种型号大孔吸附树脂中,D-101和D-101-I的回收率均低于DM-301,分析是D-101和D-101-I属于非极性树脂,对样品中的非极性杂质吸附性较强,杂质抢先占据了大孔树脂上 的吸附位点,导致部分黄芪甲苷不能吸附在大孔树脂上而随着淋洗溶液流失了。而DM-301属于中极性树脂,和黄芪甲苷的极性更为相近,对黄芪甲苷的吸附性较强,不会出现黄芪甲苷预先流失的现象。故试验选择DM-301大孔吸附树脂。
2.2 固相萃取条件的确定
基于黄芪甲苷的极性特点以及Oasis HLB固相萃取柱同时具有亲水性单体N-乙烯基吡咯烷酮和亲脂性单体二乙烯基苯,黄芪甲苷可和亲水性单体形成范德华作用力,能够充分吸附。亲脂性单体则与黄芪甲苷分子中的非极性基团作用,产生反相保留。以上两种作用使得黄芪甲苷可以保留在Oasis HLB固相萃取小柱中,当极性强的溶剂通过时可以将极性比黄芪甲苷强的物质洗脱下来,选用极性合适的溶剂将黄芪甲苷洗脱下来时,极性比它弱的物质则继续保留在固相萃取柱上。结果显示80%乙醇溶液洗脱时,黄芪甲苷回收率最高,且40%乙醇为不能洗脱下黄芪甲苷的乙醇最高浓度。故从黄芪甲苷的回收率和安全角度考虑,选择40%乙醇为淋洗液,80%乙醇为洗脱液。
2.3 衍生化反应条件的确定
试验选择苯甲酰氯和三氯甲烷作为衍生化试剂,是基于在吡啶溶液中苯甲酰氯可将黄芪甲苷中的羟基衍生为苯甲酰基,最终生成苯甲酰黄芪甲苷,使其最大紫外吸收波长发生红移,避免低紫外波长区杂质的干扰。根据试验结果结合正交设计分析方法,确定反应时间12h,反应温度4℃,吡啶用量lml,苯甲酰氯用量0.4ml为最佳衍生化反应条件。
波长扫描结果显示衍生化显色体系在 λ=230nm处有最大吸收,干扰较小。因此确定230nm波长为检测波长(图1)
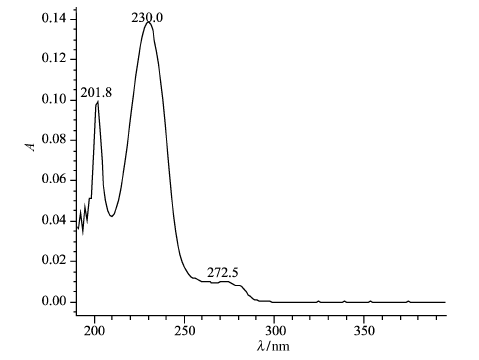
图1 黄芪甲苷衍生物的紫外光谱图
衍生化产物的质谱验证结果显示,在标准和样品的一级全扫质谱图上,均有丰度较高的1744分子离子峰,二者1744 分子离子峰的二级全扫质谱图中几个丰度较高的分子离子峰丰度比基本相同,判断二者1744 分子离子峰表示同一种物质,分析是黄芪甲苷分子结构中有10个羟基被苯甲酰化后又加上了一个钠离子,证明该法较好的消除了杂质干扰,以衍生物色谱峰面积对黄芪甲苷进行定量,准确度高(图2~5)。
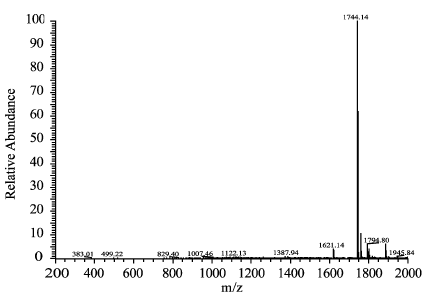
图2 黄芪甲苷标准溶液衍生化产物的一级全扫质谱图

图3 样品衍生化产物的一级全扫质谱图
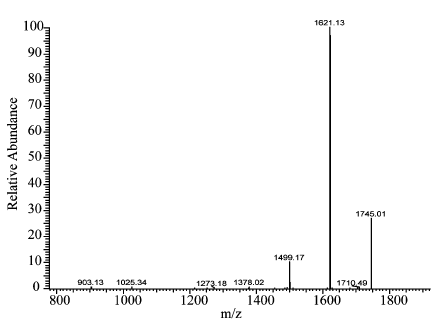
图4 黄芪甲苷标准溶液衍生化产物的二级全扫质谱图
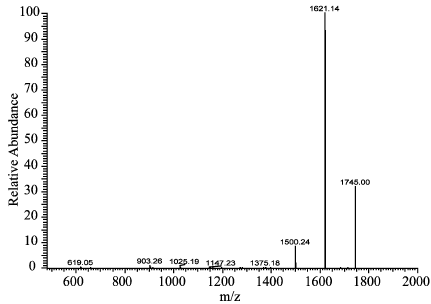
图5 样品衍生化产物的二级全扫质谱图
衍生物稳定性试验结果显示,在1小时、5小时、1周、1个 月、2个月、4个月测得的衍生物量的RSD值为1.10% ,表明衍生物在4℃下可稳定保存4个月。
2.4 高效液相色谱条件的选择
2.4.1 色谱柱的选择 结果显示Kromasil C8 保留时间在 70min左右,Nova-Pak C18, 保留时间在13min左右,二者峰形均较满意。AeeQ TagTMC18色谱峰存在一定干扰。因此选择保留时间较短、峰形较好的Novapak C18色谱柱。
2.4.2 流动相的选择 结果显示,90%甲醇-10%水未出峰, 100%乙腈时峰受干扰严重,90%甲醇-4%四氢呋喃-6%水与90%甲醇-4%四氢呋喃-6%水-0.4%三乙胺保留时间和峰形 均良好,因此选择配制简单的流动相90%甲醇一4%四氢呋喃-6% 水。
2.5 样品前处理流程和标准曲线的绘制
称取适量样品,甲醇超声提取,提取液离心取上清,水浴蒸干后用蒸馏水溶解残渣,转入分液漏斗,用无水乙醚脱脂,每次60ml,共3次,弃醚层。水层用40ml 1%水饱和正丁醇提取,共3次,弃水层。合并正丁醇层,将正丁醇层用40ml 1% KOH 洗3次,弃碱液层。将正丁醇在沸水浴上蒸干,用5ml蒸馏水溶解残渣。移入处理过的DM-301大孔树脂柱,用水和30%乙醇淋洗,80%乙醇洗脱,收集洗脱液,蒸干后用40% 乙醇溶解,移人预处理过的固相萃取柱,40%乙醇淋洗,80%乙醇洗脱,洗脱液蒸干后用lml吡啶溶解。向吡啶溶液中加入2ml 三氯甲烷和0.4ml苯甲酰氯,充分振摇lmin,4℃ 衍生12h,氮气吹干,甲醇定容。在给定仪器条件下标准溶液和样品上机检测。具体仪器条件为:色谱条件:Novapak C18 色谱柱,流动相90%甲醇-4%四氢呋喃-6%水,流速lm]/min,柱温25℃,波长230nm,进样量20μl。
精确量取0.10、0.20、0.50、1.0、2.0和4.0ml标准储备液至10.0ml容量瓶中,用甲醇定容到10.0ml,水浴蒸干后,用 1.0ml吡啶溶解后加入2.0ml三氯甲烷和0.4ml苯甲酰氯在 4℃ 下衍生12h,衍生液氮气吹干后用甲醇定容到10.0ml。进行液相色谱测定。结果线性方程为:y= -236278.71+ 76061.56X,r=0.9996。
2.6 加标回收率、精密度试验和检测限的测定
样品测定结果显示,加标回收率为74.85% ~91.83%;精密度RSD值为7.46% ;检测限为2.13 μg/ml,表明试验方法能较好满足样品测定要求。
2.7 样品的测定
按照以上建立的检测方法对多种含黄芪市售样品进行检测,结果显示峰形良好,干扰较少。两样品色谱图见图6、图7。
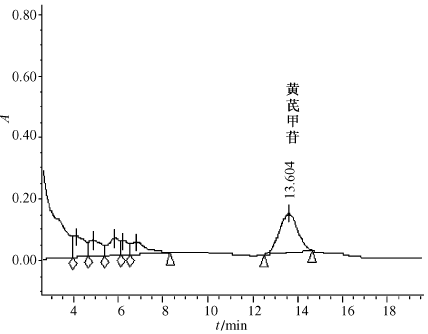
图6 样品1色谱图

图7 样品2色谱图
3 讨论
现在很多保健食品以黄芪为其主要功效成分,尚无统一有效的测定保健食品中的黄芪甲苷的方法,本研究建立了准确、灵敏、稳定的成份复杂保健食品中黄芪甲苷测定方法,对黄芪及其制剂的质量评价和合理开发利用提供了良好的实验基础。
本试验将HLB Oasis 3cc固相萃取小柱应用到富集和净化黄芪甲苷的前处理步骤中,结果证明,此固相萃取小柱净化效果良好,对保健食品中黄芪甲苷的除杂和富集必不可少。本试验首次将DM-301大孔吸附树脂应用到黄芪甲苷的前处理除杂步骤中,按照极性由大到小的顺序进行淋洗,依次除去样品中的各种极性杂质。大孔树脂化学性质稳定,不溶于酸、碱及有机溶剂,对有机物有浓缩分离作用且不受无机盐类及强离子、低分子化合物的干扰,操作简便,因此大孔树脂在保健食品的除杂过程中是一种较理想的手段。
黄芪甲苷在近紫外区仅仅200nm末端有吸收,且灵敏度很低,黄芪含有的多种成分对结果干扰很大。本试验采用柱前衍生化-高效液相色谱法,对黄芪甲苷分子中的羟基进行苯甲酰化反应,使最大吸收波长红移至230nm处,提高了分离选择性和检测灵敏度,大大减少了杂质的干扰。最低检出限、加标回收率、精密度均较好。因此,本试验建立了成分复杂保健 食品中黄芪甲苷的柱前衍生化-高效液相色谱测定法,为制定保健食品黄芪甲苷标准测定方法提供了较好的方法基础。